Research Topic
Data-Driven Ligand Optimization
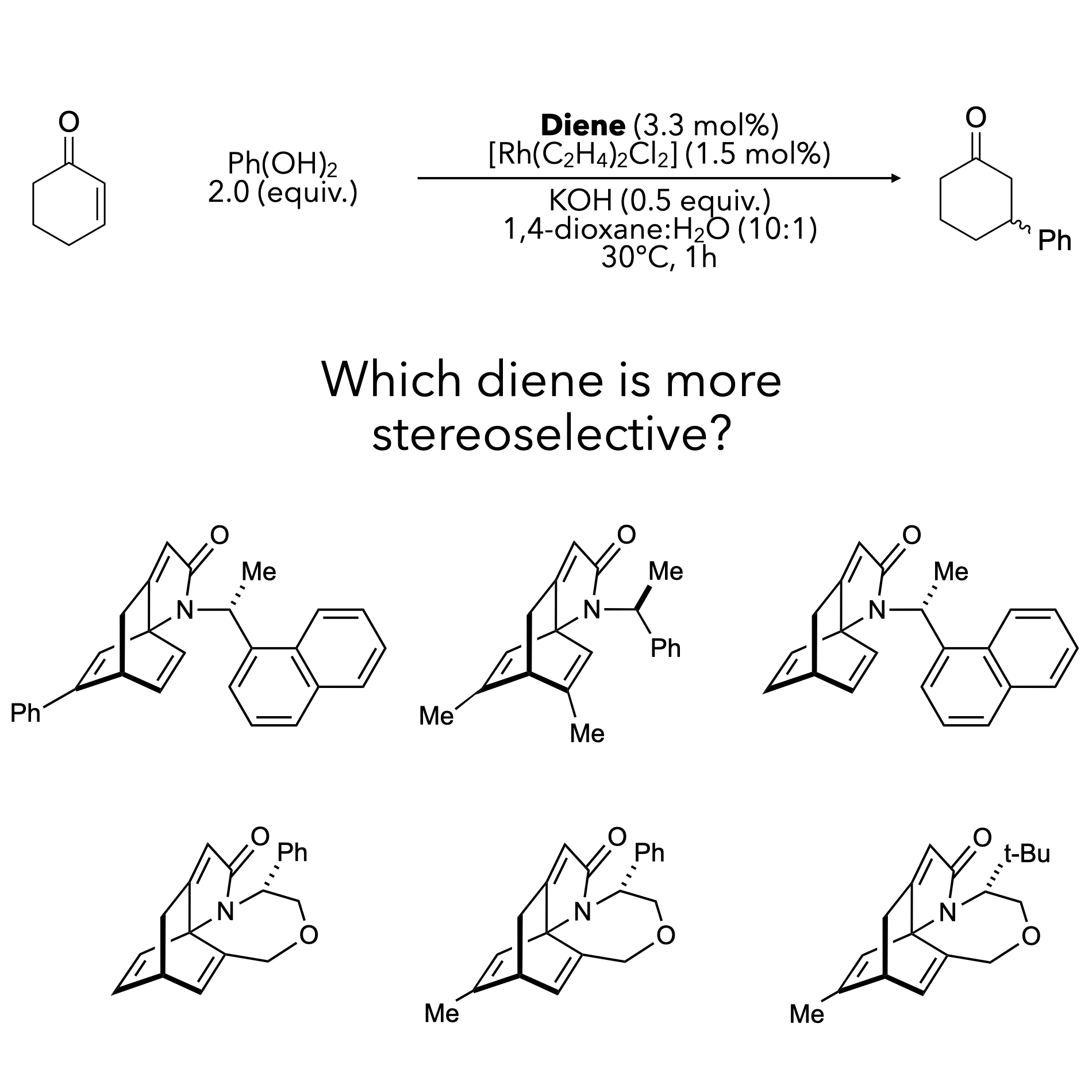
Traditional approaches to ligand design involve synthesising numerous structural variants — a process that is time-consuming and expensive. We aim to leverage machine learning to make this process dramatically more efficient.
Asymmetric Catalysis
Graph Neural Networks
Explainable AI
Cheminformatics